CDK2 Clustering¶
- The simplest way to cluster structures based upon residue properties is to use the polyphony_Ete2_tree.py script. This will generate an interactive tree in Ete and write out a Newick format file. This is a standard tree text format and can be read in using a number of programs. Here is a dendrogram produced using FigTree. It shows the CDK2 chains clustered by Tanimoto similarity of the residue interaction fingerprints generated from protein-protein interaction database Piccolo (-p ppi option).
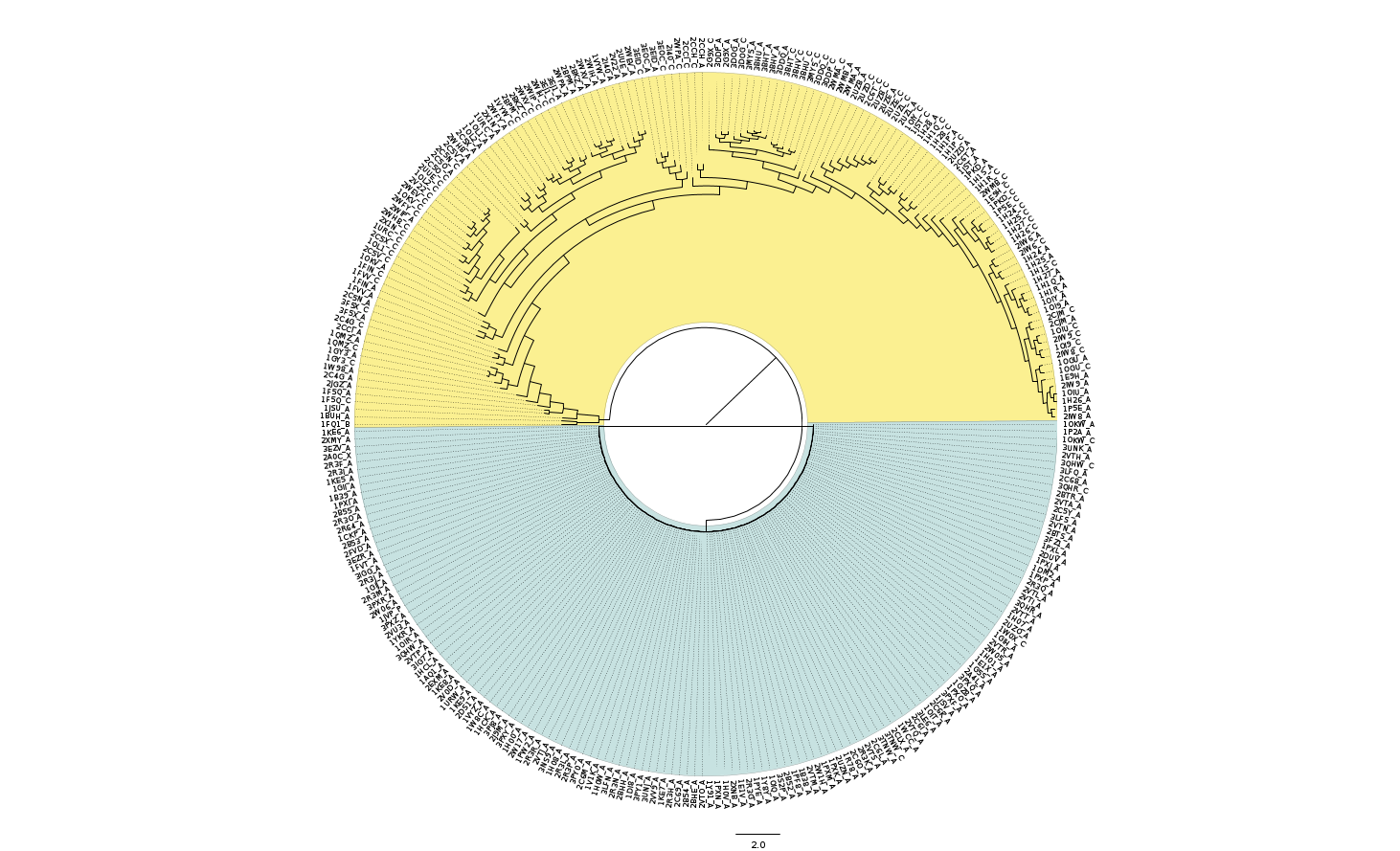
It shows the chains with no biologically relivant protein contacts in blue and those interacting, usually with cyclin a, in yellow.
- Groups can be generated from the hierarchical clustering. This isn’t done in a very sophisticated way. One way is to use:
This method works well to split CDK2 chains into monomers and multimers (blue and yellow in above diagram).
e.g.
from Polyphony.Trees import Tree
from Polyphony.Comparison_Matrices import Structure_Matrix
from Polyphony.Structural_Alignment import Structural_Alignment
from Polyphony.Utils import Properties
from Polyphony.Plotting import plot_alignment_properties
## Main program
# Alignment file etc.
filename = "clust_1HCK_A_95.fasta"
update = False
property = "backbone"
# Create structural alignment
aligned = Structural_Alignment()
aligned.add_alignment(filename)
# Get/calculate selected property
properties = Properties()
array = properties.get_array(property, aligned, update)
# Cluster by protein-protein interactions
clust_array = properties.get_array("ppi", aligned, update)
structmat = Structure_Matrix(clust_array)
tree = Tree(structmat.data, structmat.get_labels())
# Group into active and inactive
groups = tree.biggest_left_right_others(1)
unbound = groups[0]
bound = groups[1]
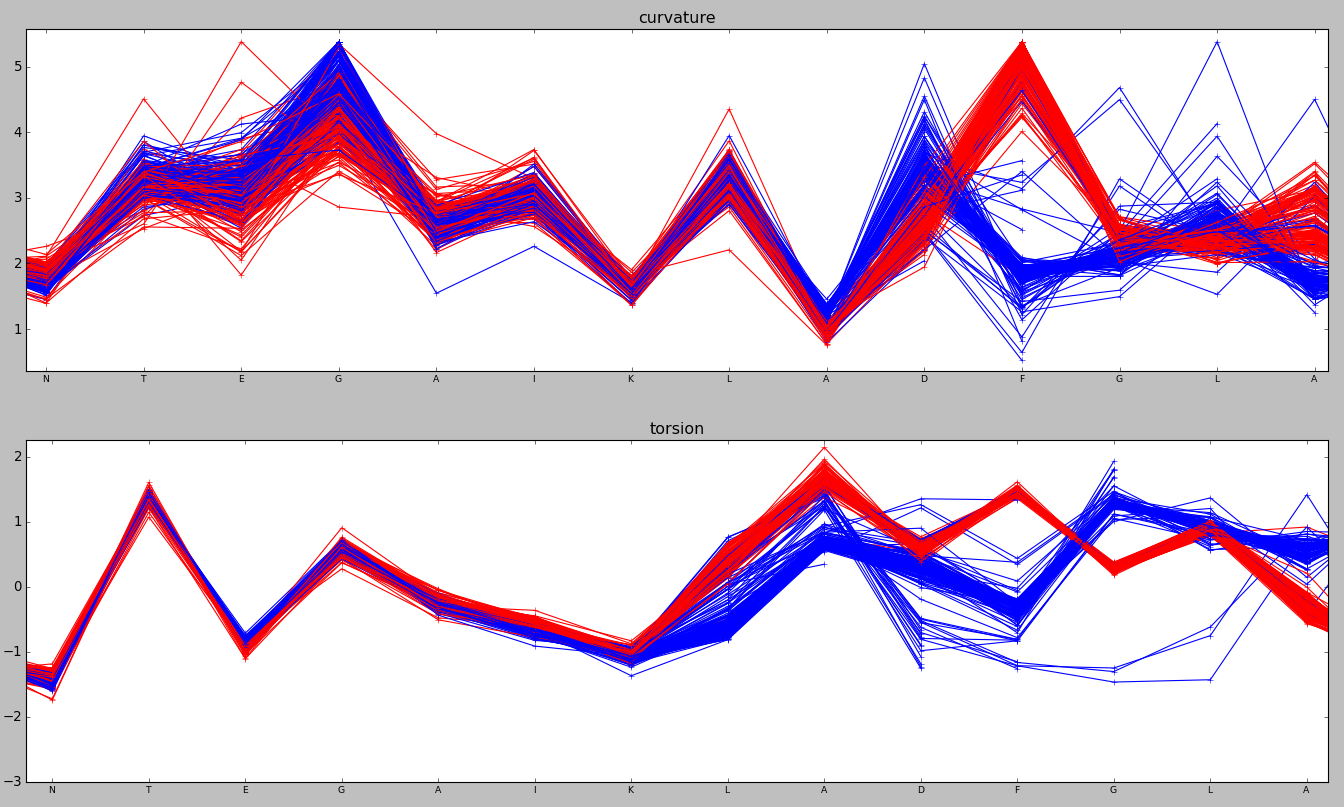
# Plot curvature and torsion, coloured by group
xlabels = aligned.get_consensus_sequence()
plot_alignment_properties(array.data, property, xlabels, array.dim_names, colour_groups=[unbound, bound])
The same region can be visualised in 3D using in PyMol. Type pymol -R in one console window and run the following in another, preferably in ipython.
from Polyphony.Pymol import Pymol_Viz
# Get started and load representative structure into PyMol
cdk2 = Pymol_Viz("clust_1HCK_A_95.fasta","cdk2")
# Group in to bound and unbound structures
groups = cdk2.group_biggest_clusters(property="ppi")
unbound = groups[0]
bound = groups[1]
# Convert the lists of alignment index numbers to chain ids
unbound_ids = cdk2.ids_for_groups(unbound)
bound_ids = cdk2.ids_for_groups(bound)
# Find preferred segment of structurally conserved residues. Try experimenting with the cutoff. The default value of 0.5 can be a bit high.
cdk2.colour_conserved_segments(0.1)
# Load all structures in each group into a PyMol group allowing them to be coloured separately
# They then aligned using your chosen segment. This will take a few minutes
cdk2.load_structures(id_list=unbound_ids, segment=[101,111], pymol_group="unbound")
cdk2.load_structures(id_list=bound_ids, segment=[101,111], pymol_group="bound")
You can then colour each group separately, view as ribbons etc.
