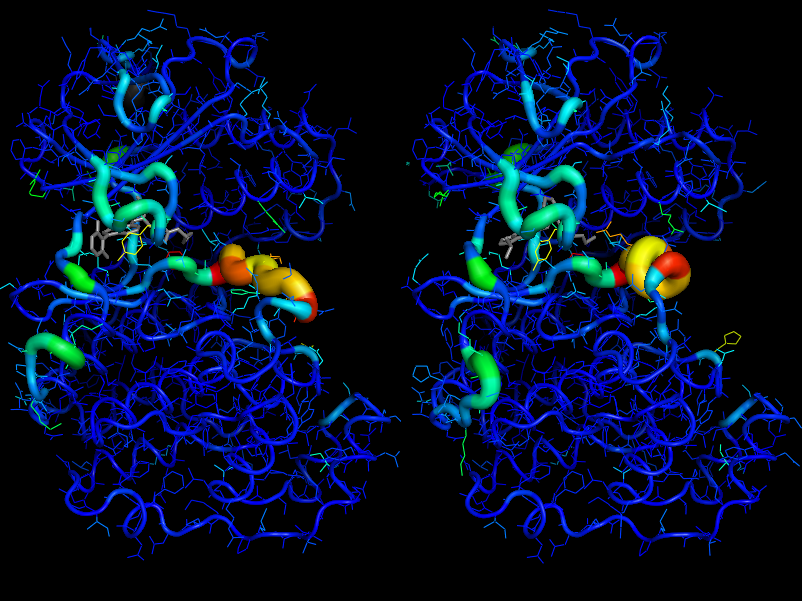
Example PyMol API output¶
Here are shown the output generated for the cdk2.fasta alignment file in the examples directory using the PyMol API.
from Polyphony.Pymol import Pymol_Viz
cdk2 = Pymol_Viz("cdk2.fasta","cdk2")
cdk2.colour_by_variability()
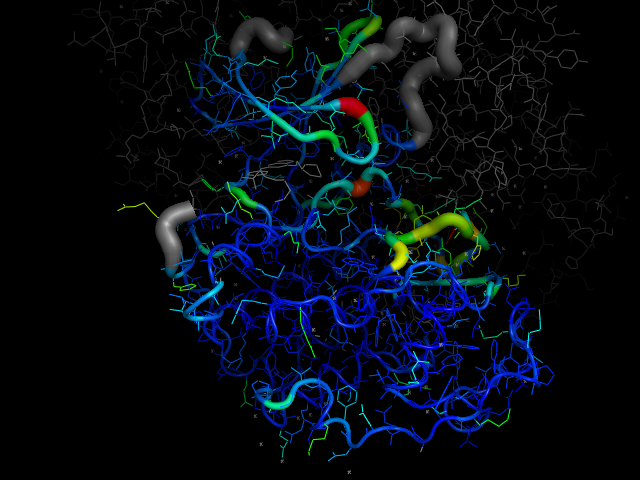
cdk2.colour_by_average_bfactor()
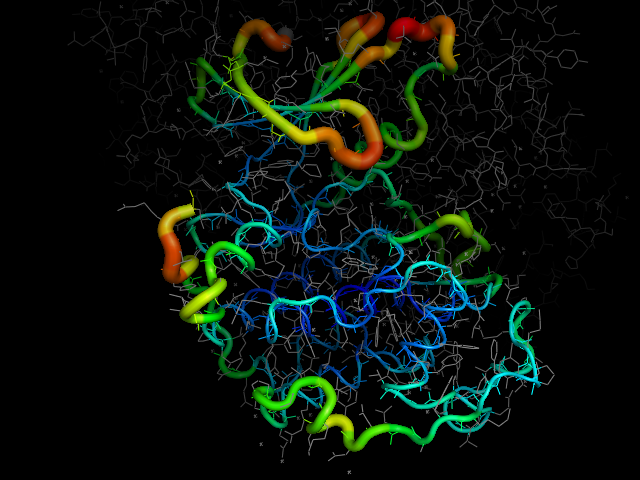
cdk2.colour_by_seq_variability()
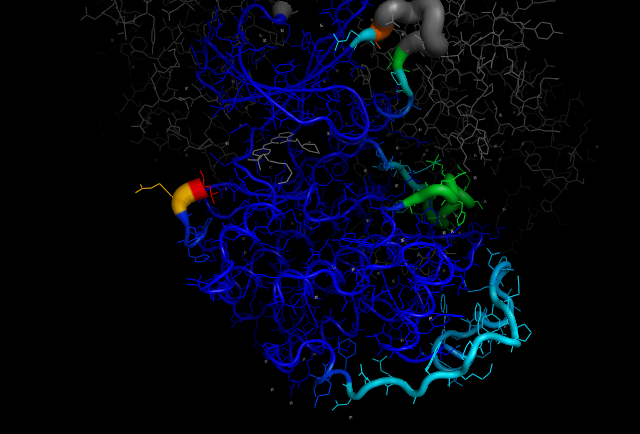
cdk2.colour_by_Ramachandran_region()
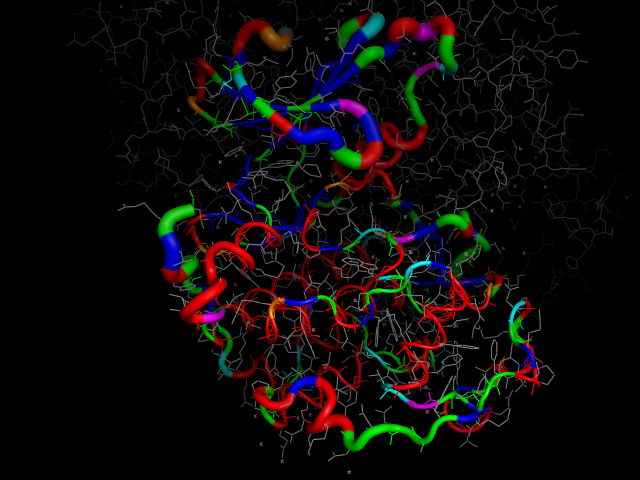
cdk2.colour_by_contacts(type="crystal_contacts")
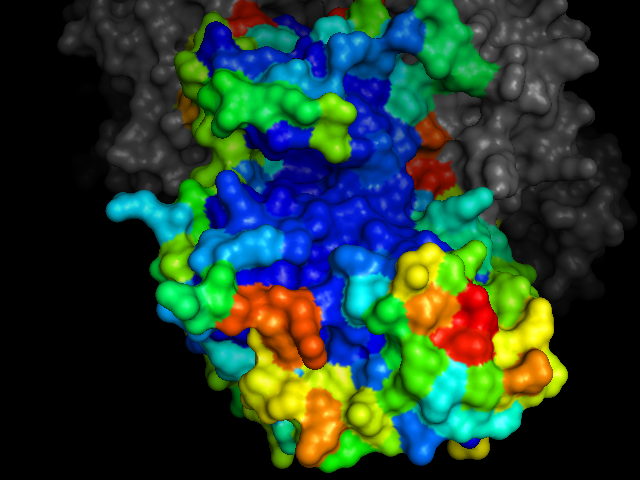
cdk2.colour_by_pockets()
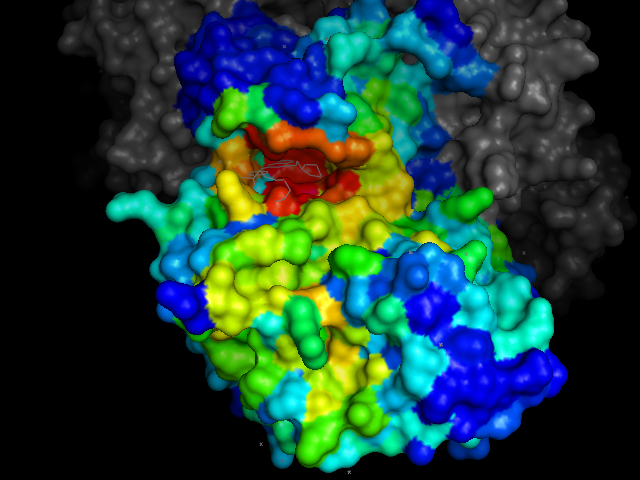
cdk2.colour_by_druggable_pockets()
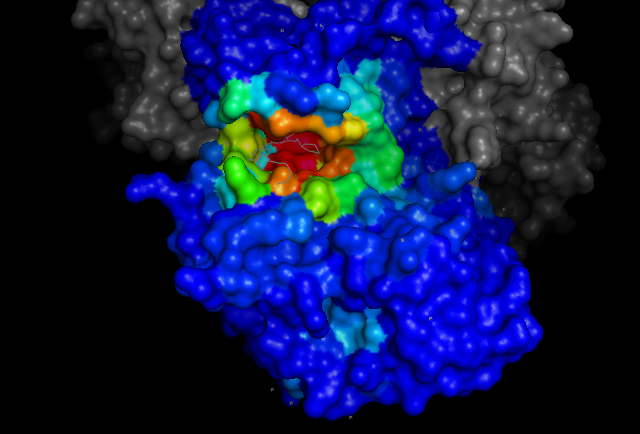
cdk2.colour_by_pca(max_pc_chains_to_ignore=15, components=[0])
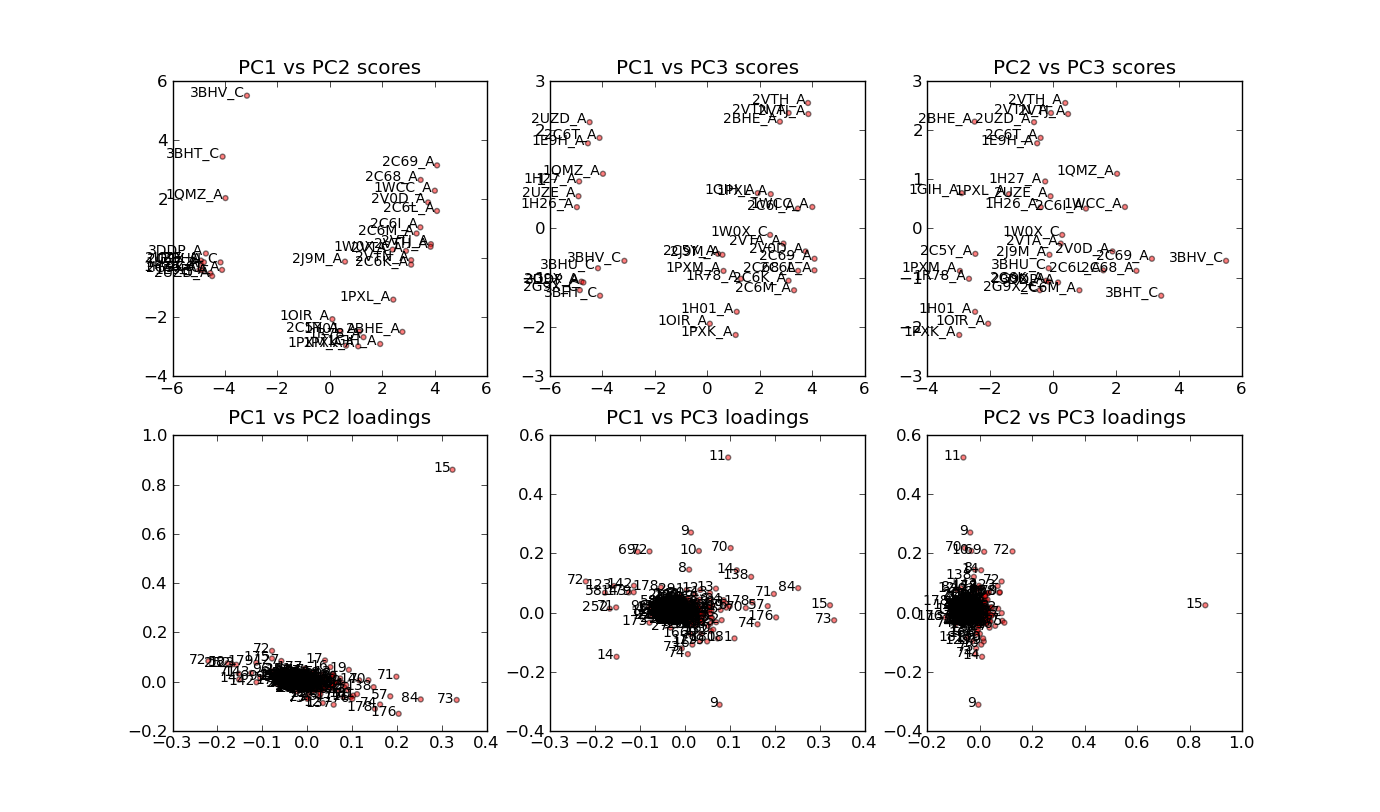
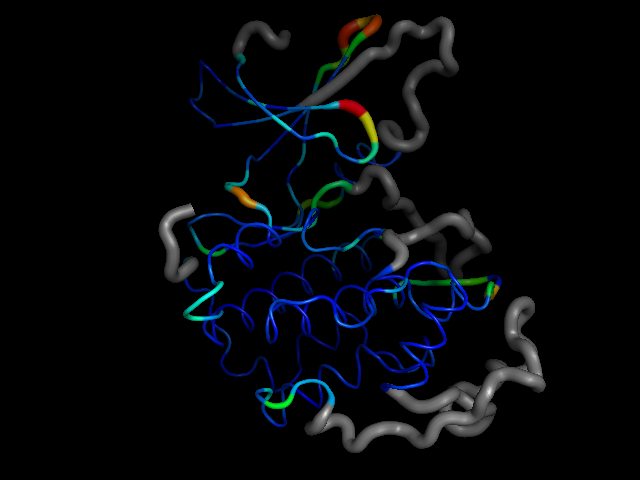
cdk2.colour_conserved_segments(20, separation=50)
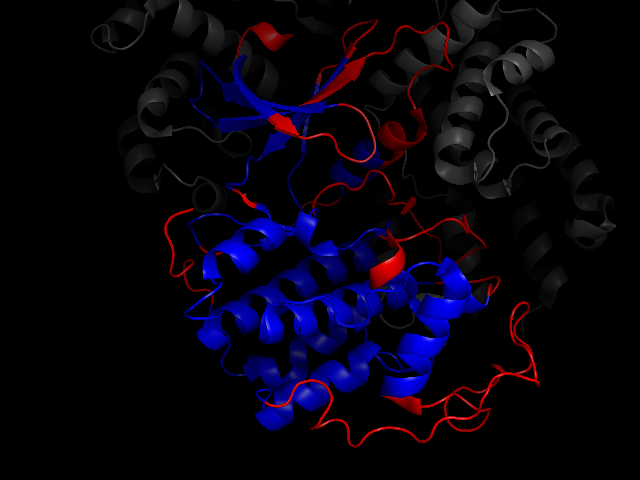
Show the 20 residue pairs, separated by at least 50 residues, with most correlated backbone conformations.
cdk2.show_correlated_backbones(20, separation=50)
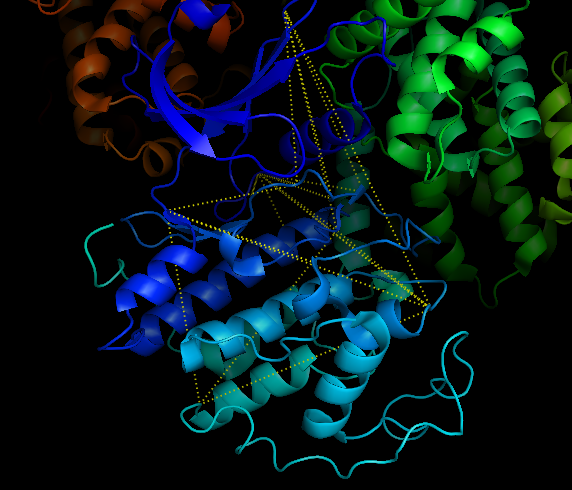
Show the 50 residue pairs, separated by at least 5 residues, with most correlated side-chain conformations.
cdk2.show_correlated_sidechains(50, separation=5)

Show backbone regions and sidechains that change conformation when cyclin is bound to cdk2
from Polyphony.Pymol import Pymol_Viz
cdk2 = Pymol_Viz("clust_1H26_A_90.fasta","cdk2")
#
# group by Piccolo annotated protein-protein interactions
groups = cdk2.group_biggest_clusters(property="ppi")
active = groups[0]+groups[1]
inactive = groups[2]
#
# default structure is without cyclin bound
cdk2.colour_by_amplification(property="backbone", groups=[active,inactive])
cdk2.colour_by_amplification(property="sidechain", groups=[active,inactive])
#
# load structure representative of cyclin bound state
active_rep = cdk2.get_representative_structure([active])
cdk2.load_structures([active_rep])
cdk2.colour_by_amplification(property="backbone", groups=[active,inactive], chain_id=active_rep)
cdk2.colour_by_amplification(property="sidechain", groups=[active,inactive], chain_id=active_rep)
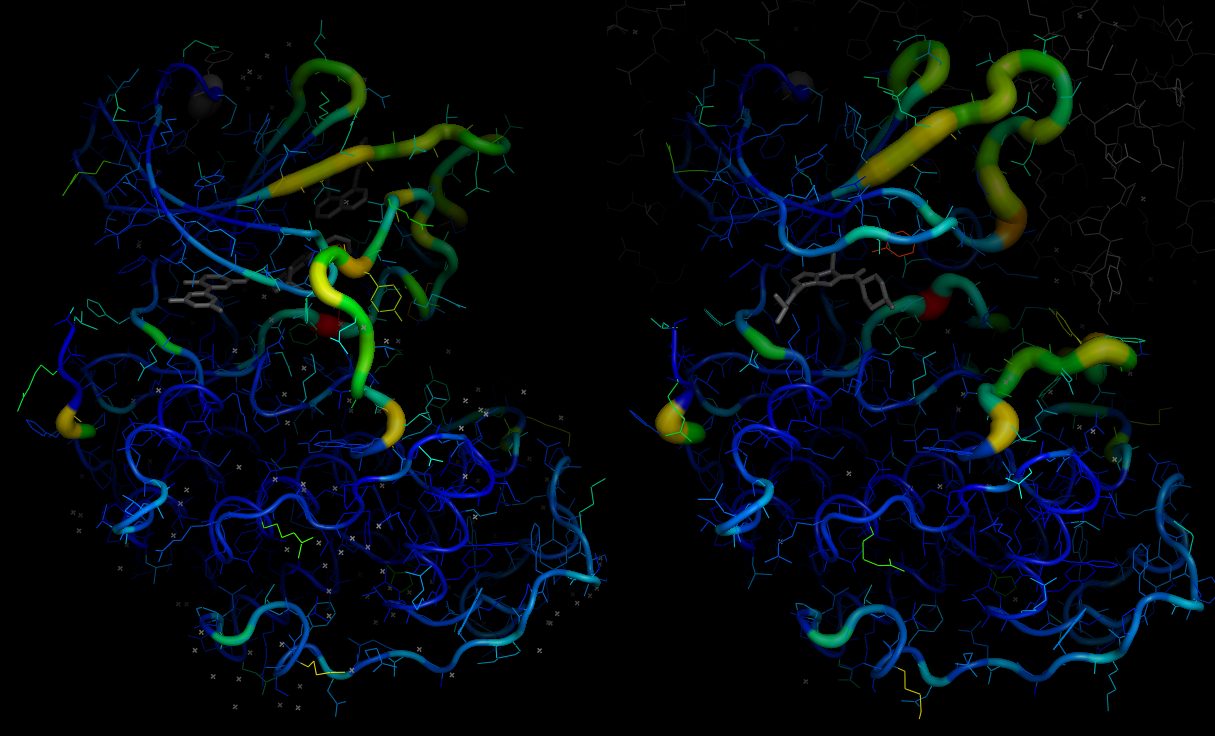
Do similar for p38, after clustering by backbone conformation
# Read in alignment
p38 = Pymol_Viz("../p38/clust_1A9U_A_90.fasta","p38")
# Cluster by backbone conformation in 2 largest groups [0,1] and then into largest subgroups [2,3] and [4,5], [6] contains outliers
p38_groups = p38.group_biggest_clusters(3)
p38_group_names = p38.ids_for_groups(p38_groups)
p38_b1 = p38_groups[0]
p38_b2 = p38_groups[1]
p38_b11 = p38_groups[2]
p38_b12 = p38_groups[3]
p38_b21 = p38_groups[4]
p38_b22 = p38_groups[5]
# Load representative structure (most ordered residues) for each group into PyMol
p38_b1_rep = p38.get_representative_structure([p38_b1])
p38_b2_rep = p38.get_representative_structure([p38_b2])
p38_b11_rep = p38.get_representative_structure([p38_b11])
p38_b12_rep = p38.get_representative_structure([p38_b12])
p38_b21_rep = p38.get_representative_structure([p38_b21])
p38_b22_rep = p38.get_representative_structure([p38_b22])
# load representative structure aligned to residues 127-144 for consistency with cdk2 example
p38.load_structures([p38_b1_rep,p38_b2_rep,p38_b11_rep,p38_b12_rep,p38_b21_rep,p38_b22_rep], segment=(127,144))
# Colour residues by amplified changes between subgroups b21 and b22
p38.colour_by_amplification(property="backbone",groups=[p38_b21,p38_b22],chain_id=p38_b22_rep)
p38.colour_by_amplification(property="backbone",groups=[p38_b21,p38_b22],chain_id=p38_b21_rep)
p38.colour_by_amplification(property="sidechain",groups=[p38_b21,p38_b22],chain_id=p38_b21_rep)
p38.colour_by_amplification(property="sidechain",groups=[p38_b21,p38_b22],chain_id=p38_b22_rep)