4. Creating Jalview Feature files¶
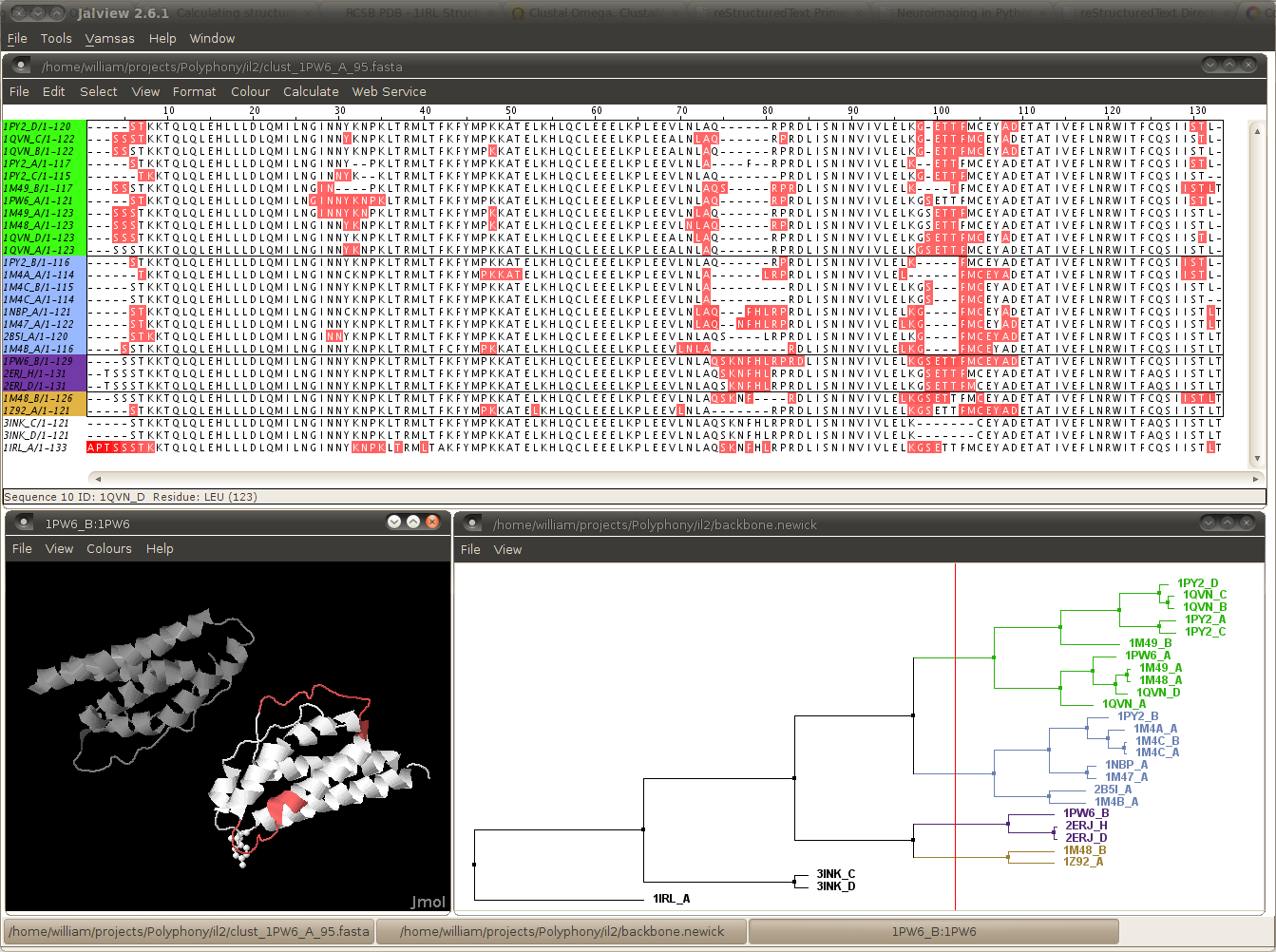
Jalview is a very powerful sequence alignment viewer and editor. See Optional Add-ons for more info. Here we will create a Jalview feature file for highlighting residues in the alignment and a dendrodram which can be used so sorting the sequences in an alignment.
polyphony_write_jalview_feature_file.py -f clust_1PW6_A_95.fasta -p bfactor
Generates a file called bfactor.ano. This is a Jalview feature file which can be used to highlight residues with high temperature factors in a sequence alignment view.
polyphony_write_newick.py -f clust_1PW6_A_95.fasta
Creates a file called backbone.newick. This is tree representation of the a clustering by backbone conformation. Use the -p option to specify alternative properties to use for the clustering.
Both these files can be read into Jalview like so:
Start Jalview
File > Input Alignment > from File (read in your fasta file e.g. clust_1PW6_A_95.fasta)
- A sequence alignment viewer window should open in that window
- File > Load Features/Annotations (read in bfactor.ano)
- File > Load Associated Tree (read in backbone.newick)
- Calculate > Sort > By Tree Order > /your_path/backbone.newick
You can now click on the tree view to colour sequences by cluster. You can also view the feature colours in 3D by e.g.
- right click on a structure label in the alignment view (e.g. 1PW6_B) > Structure > Associate Structure with Sequence > Enter PDB id > 1PW6
- right click on same structure label in the alignment view > Structure > View Structure > 1PW6
The results should look something like this (after a bit of fiddling with windows and fonts etc.)